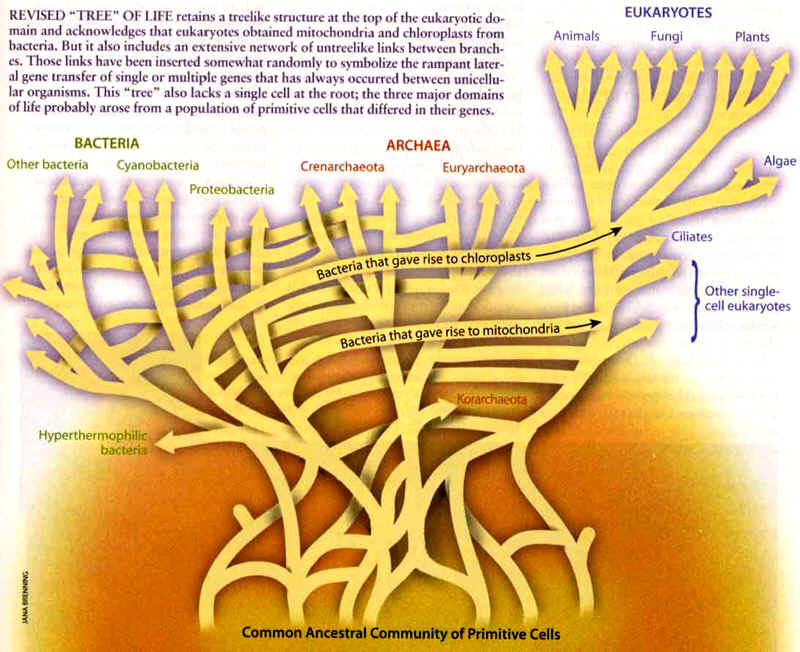
For many evolutionary biologists, the most significant single piece of evidence supporting the Darwinian theory of origins is the nested hierachical pattern that is formed when comparing various genetic sequences in different organisms. The similarities and differences, it is argued, map out into a kind of "Tree of Life". It is this consistent pattern created by numerous different genes and genetic sequences that appears to many to be extremely compelling evidence.
For example, in the video above Richard Dawkins was asked, "Out of all the evidence used to support the theory of evolution, what would you say is the strongest, most irrefutable single piece of evidence in support of the theory?" (Link). Dawkins' response is most interesting:
.
"I think to me perhaps the most compelling evidence is comparative evidence, from modern animals -- particularly biochemical comparative evidence, genetic, molecular evidence.
If you take any set of animals, and identify the same gene in different animals, and you really can do that, because the letters of the DNA code -- that is, the same code in all animals -- and you really can find a gene which is the same -- in, say, all mammals. For example, there's a gene called FOXP2, which is a couple of thousand letters long, and most of the letters are the same in any mammal, so you know it's the same gene. And then you go through, and you literally count the number of letters that are different.
So, in the case of FOXP2, if you count the number of letters that are different between humans and chimpanzees, it's only about 9. If you count the number of letters that are different in humans and mice, it's, I don't know, 30 or something like that. Actually, frogs have them as well, you find a couple of hundred that are different.
So, you can take any pair of animals you like -- kangaroo and lion, horse and cat, human and rat -- any pair of animals you like, and count the number of differences in the letters of a particular gene, and you plot it out, and you find that it forms a perfect branching hierarchy.
It's a tree, and what else could that tree be, but a family tree. And then you do the same thing for another gene. Having got the family tree for FOXP2, you then do the same thing for another gene, and another, and another. You get the same family tree.
You also get the same family tree if you take genes that are no longer functioning, that are just vestigial, that are not doing anything. It's like fragments of a document on your hard disk, which are no longer being used, they're no longer on the directory, so you no longer see them. Again, you get the same family tree.
This is overwhelmingly strong evidence. The only way you could get out of saying that that proves evolution is true is by saying that the intelligent designer, God, deliberately set out to lie to us, deliberately set out to deceive us." (Link, Link).
.
.
The problem with this claim is, of course, is that more and more inconsistent phylogenies are being discovered and more and more genes and genetic sequences are studied. And, quite surprisingly for many scientists, many of the phylogenies based on these various genetic sequences are very inconsistent with each other and with standard Darwinian ideas based on morphologic classification models. In fact, as detailed below, the entire "Tree of Life" concept is starting to be seriously questioned by many very well known scientists who are starting to recognize such common and often dramatic inconsistencies between various phylogenic "trees".
.
.
Problems with RNA:
Current animal and plant classification models are fairly subjective in how they are set up. Scientists had hoped that the newer science of molecular biology would provide more objectivity to classification systems.It was hoped that comparisons of the nucleotides of DNA or RNA sequences or of amino acid sequences in proteins would yield more consistent results that could be used to classify organisms with a high degree of accuracy.However, quite some time ago (January 1998 issue of Science) it was discovered that this notion hope didn't play out in real life:
Animal relationships derived from these new molecular data sometimes are very different from those implied by older, classical evaluations of morphology. Reconciling these differences is a central challenge for evolutionary biologists at present. Growing evidence suggests that phylogenies of animal phyla constructed by the analysis of 18S rRNA sequences may not be as accurate as originally thought. Inaccuracies may occur in molecular phylogenies for a variety of reasons.
Prior to analysis, the sequences of corresponding genes from each animal must be placed in register (aligned) with each other so that homologous sites within each sequence can be compared. However, sequence divergences may be sufficiently large that unambiguous alignments cannot be achieved, and different alignments may lead to different inferred relationships. Additionally, the data are often sufficiently noisy that there may be a lack of strong statistical support for important groupings. 1
The article goes on to present similarities and differences in 18s rRNA sequences which show that mollusks (scallops) are more closely related to deuterostomes (sea urchins) than arthropods (brine shrimp). Of course, this is not too surprising.Intuitively, a scallop seems more like a sea urchin than a shrimp.So, the 82% correlation between the scallop and sea urchin is not surprising. However, in this light it is surprising is that a tarantula (also an arthropod) has a 92% correlation with the scallop. Here we have two different arthropods, a shrimp and an tarantula. How can a scallop be much more related to one type of arthropod and much less related to the other type of arthropod? This troubling thought led the authors of the Science article to remark:
Different representative species, in this case brine shrimp or tarantula for the arthropods, yield wildly different inferred relationships among phyla. Both trees have strong bootstrap support (percentage at node). . . The critical question is whether current models of 18S rRNA evolution are sufficiently accurate to successfully compensate for long branch attraction between the animal phyla. Without knowing the correct tree ahead of time, this question will be hard to answer. However, current models of DNA substitution usually fit the data poorly. . . 1
There are many other interesting little problems concerning commonly used phylogenic tracing genes and proteins - problems which have been known for some time now. For example, mammalian and amphibian "luteinizing hormone - releasing hormone" (LHRH) is identical. However, birds, reptiles, and certain fish have a different type of LHRH.Are humans therefore more closely related to frogs than to birds?Not according to standard evolutionary phylogeny trees. Again, the data does not match the classical theory in this particular situation.15
Calcitonin (lowers blood calcium levels in animals) is another protein commonly used to determine phylogenies. Interestingly though humans differ from pigs by 18 of 32 amino acids, but by only 15 of 32 amino acids from the salmon.Are we therefore more closely related to fish than to other mammals like the pig? 5
Cytochrome c is another famous phylogenic marker protein used to determine evolutionary relationships.There are no sequence differences between humans and chimps and only one difference between human and rhesus monkey cytochrome c sequences.Because of this, many assume that the evolutionary link is obvious.However, with many other animals, this link is not so obvious.For example, the cytochrome c protein of a turtle is closer to a bird than it is to a snake and a snake is closer to a human (14 variations) than it is to a turtle (22 variations) - (Ambler and Daniel, 1991).5 How is this explained? By "convergent evolution", but without an apparent reason for such convergence:
.
"We believe that the sequence anomaly [i.e., a closer relationship between snakes and humans vs. snakes and turtles] is best explained by there having been rapid evolution in the line leading to the Colubridae, and that some convergence of the primate and this snake line has occurred. We can think of no rationale to explain such convergence for these cytochromes c, as has been adduced for the stomach lysozymes of ruminants and leaf-eating primates (Stewart et al., 1987), where the convergent evolution of a fermentative foregut in two groups of mammals appears to be complemented by convergence in their bacteriolytic lysozymes." (Ambler and Daniel, 1991).
.
Humans and horses, both being placental mammals, are presumed to have shared a common ancestor with each other more recently than they shared a common ancestor with a kangaroo (a marsupial). So the evolutionist would expect the cytochrome c of a human to be more similar to that of a horse than to that of a kangaroo. Yet, the cytochrome c of the human varies in 12 places from that of a horse but only in 10 places from that of a kangaroo.5
Again, such discrepancies between traditional phylogenies and those based on cytochrome c are well known and have been well known for a long time now. Ayala commented that:
.
"The cytochrome c phylogeny disagrees with the traditional one in several instances, including the following: the chicken appears to be related more closely to the penguin than to ducks and pigeons; the turtle, a reptile, appears to be related more closely to birds than to the rattlesnake, and man and monkeys diverge from the mammals before the marsupial kangaroo separates from the placental mammals." 8
.
Even so, cytochrome c does seem to generally match the predictions of common decent. However, there are some who think that the general cytochrome c data presents some puzzles from a neo-Darwinian perspective. First, the cytochromes of all the higher organisms (yeasts, plants, insects, fish, amphibians, reptiles, birds, and mammals) exhibit an almost equal degree of sequence divergence from the cytochrome of the bacteria Rhodospirillum. In other words, the degree of divergence does not increase as one moves up the scale of evolution but remains essentially uniform. The cytochrome c of other organisms, such as yeast and the silkworm moth, likewise exhibits an essentially uniform degree of divergence from organisms as dissimilar as wheat, lamprey, tuna, bullfrog, snapping turtle, penguin, kangaroo, horse, and human. 5, 6 According to Michael Denton, a molecular biology researcher, "At present, there is no consensus as to how this curious phenomenon can be explained." 7
What is really interesting, however, However, is that evolutionists cherry pick the Cytochrome C example while rarely talking about the Cytochrome B tree, which has striking differences from the classical animal phylogeny. As one article in Trends in Ecology and Evolution stated:
.
"The mitochondrial cytochrome b gene implied... an absurd phylogeny of mammals, regardless of the method of tree construction. Cats and whales fell within primates, grouping with simians (monkeys and apes) and strepsirhines (lemurs, bush-babies and lorises) to the exclusion of tarsiers. Cytochrome b is probably the most commonly sequenced gene in vertebrates, making this surprising result even more disconcerting." 28
So, the data does seem to generally match the theory even if specific anomalies may be encountered on relatively "rare" occasions in such cases as cytochrome c phylogenies. However, there might be a few problems with this scenario definitively supporting the theory of common decent. One problem might arise when one considers that mutation rates are calculated on a per generation average. Consider that the average mutation rate for a given gene in all creatures, is about 1 x 10-6 mutations per gene per generation. That means that a given gene will mutate only one time in one million generations on average. Consider that single celled organisms have a much shorter generation time than multi-celled organisms on average. For example, the bacteria E. coli have a minimum generation time of 20 minutes compared to the generation time of humans of around 20 years. With a gene being mutated every 1 to 10 million generations in E. coli, one might think this would be a long time. However, each and every gene in an E. coli lineage will get mutated once every 40 to 80 years.So, in one million years, each gene will have suffered at least 10,000 mutations.
Now, cytochrome c phylogenies are generally based on analysis of certain subunits of cytochrome c which range in number of amino acids up to a maximum of about 600 or so. This would translate into a minimum of at least 1,800 nucleic acids in DNA coding for this subunit of cytochrome c protein (3bp per codon). Note that in the table above, the tetrahymena species are about 50% different from all other creatures on the table. It seems then that all the creatures would have experienced at least a 25% change in their genetic codes from the time of common ancestor. So how many generations would it take to achieve this 25% difference?
Taking 25% of 1,800 give us 450 mutations. Lets say that the average mutation rate is one mutation per 1,800 nucleic acids per one million generations. For a steady state population of just one individual in each generation it would take about 450 million generations to get a 25% difference from the common ancestor. With a generation time of 20 minutes (ie: E. coli), that works out to be about 342,000 years. So, for bacteria, the 25% difference from the common ancestor cytochrome c, might have been achieved relatively rapidly given the evolutionary time frame. (See additional addendum comments below)
The question is, then, if bacteria can achieve such relatively rapid neutral genetic drift, why are they not more wide ranging in their cytochrome c sequences? It seems that if these cytochrome c sequence differences were really neutral differences, that various bacterial groups, colonies, and species, would cover a rather large range of possible cytochrome c sequences - potentially to include that of mammals. Why are they then so uniformly separated from all other "higher" species unless the different cytochrome sequences are different for functional reasons? that they are statically different due to the various different functional needs of creatures that inhabit different environments?
For example, bacteria are thought to share a common ancestor with creatures as diverse as snails, sponges, and fishes. The split from the common ancestry of the creatures is thought to have happened over 3 billion years ago. Then, about 600 million years ago there was the Cambrian explosion where all the major phyla of living things are thought to have suddenly evolved. So, obviously all of these creatures have all been around long enough and are diverse enough to exhibit quite a range in cytochrome c variation. Why then are their cytochrome c sequences so clustered and arranged in such an orderly hierarchy? Why don't bacteria, snails, fish, and sponges cover a more random range of cytochrome c sequence variation if these variation possibilities are in fact neutral?
I propose that the clustered differences that are seen in genes and protein sequences, such cytochrome c, are the result of differences in function that actually benefit the various organisms according to their different individual needs. If the differences were in fact neutral differences, there would be a vast overlap by now with complete blurring of species' cytochrome c boundaries - even between species as obviously different as humans and bacteria. Because of this, sequence differences may not be so much the result of differences due to random mutation over time as they are due to differences in the functional needs of different creatures. I think that the same can be said of most if not all phylogenies that are based on genotypic differences between all living things.
For example, consider that if either humans or bacteria would be better served by a different sequence for a particular function this different sequence would be rapidly evolved - especially in bacteria. If the human sequence for cytochrome c would better serve E. coli bacteria than their current fairly similar type of cytochrome c, how can an evolutionist say that E. coli would have very much trouble at all evolving the human sequence?The fact that the sequences remain consistently different over a significant span of real time observation (over a million generations for bacteria at least) is very good evidence that the differences in DNA character sequencing are based in differences of functional need, not evolutionary heritage.
Then, there is also the argument that the nested hierarchical pattern, by itself, supports the theory of common descent - even if it is known that intelligent design had to have been involved. Regardless of the involvement of intelligent design or not, the simple presence of the pattern is argument enough to support the theory of common descent - or is it? To see an interesting exchange between John Harshman and myself see the following ( Link ).
Another interesting question concerns the notion that a nested hierarchical pattern is present throughout the tree of life. This doesn't seem to be the case. It seems as though the roots of the tree do not show a nested pattern. This means that the evolutionary theory has to be able to explain both nested and non-nested patterns in the tree of life.
In this line consider the fairly recent comments from Elizabeth Pennisi in a 1999 Science article entitled, "Is it Time to Uproot the Tree of Life?"
"A year ago, biologists looking over newly sequenced genomes from more than a dozen microorganisms thought these data might support the accepted plot lines of life's early history. But what they saw confounded them. Comparisons of the genomes then available not only didn't clarify the picture of how life's major groupings evolved, they confused it. And now, with an additional eight microbial sequences in hand, the situation has gotten even more confusing . . . Many evolutionary biologists had thought they could roughly see the beginnings of life's three kingdoms . . . When full DNA sequences opened the way to comparing other kinds of genes, researchers expected that they would simply add detail to this tree. But "nothing could be further from the truth," says Claire Fraser, head of The Institute for Genomic Research (TIGR) in Rockville, Maryland. Instead, the comparisons have yielded many versions of the tree of life that differ from the rRNA tree and conflict with each other as well . . . " 10
.
Such problems were not completely unexpected. Earlier, in 1993, Patterson, Williams, and Humphries, scientists with the British Museum, reached the following conclusion in their review of the congruence between molecular and morphologic phylogenies:
As morphologists with high hopes of molecular systematics, we end this survey with our hopes dampened. Congruence between molecular phylogenies is as elusive as it is in morphology and as it is between molecules and morphology. . . . Partly because of morphology’s long history, congruence between morphological phylogenies is the exception rather than the rule. With molecular phylogenies, all generated within the last couple of decades, the situation is little better. Many cases of incongruence between molecular phylogenies are documented above; and when a consensus of all trees within 1% of the shortest in a parsimony analysis is published structure or resolution tends to evaporate.2
Also, in 1997, Hasegawa, et al, wrote:
.
That molecular evidence typically squares with morphological patterns is a view held by many biologists, but interestingly, by relatively few systematists. Most of the latter know that the two lines of evidence may often be incongruent. 27
.
,
In 1998 biologist Carl Woese, who was an early pioneer in producing rRNA-based phylogenetic trees, concluded:
"No consistent organismal phylogeny has emerged from the many individual protein phylogenies so far produced.
Phylogenetic incongruities can be seen everywhere in the universal tree, from its root to the major branchings within and among the various taxa to the makeup of the primary groupings themselves. Yet there is no consistent alternative to the rRNA phylogeny, and that phylogeny is supported by a number of fundamental genes... For example... different (related) aaRSs root that tree differently...
Exceptions to the topology of the rRNA tree such as these are sufficiently frequent and statistically solid that they can be neither overlooked nor trivially dismissed on methodological grounds. Collectively, these conflicting gene histories are so convoluted that lateral gene transfer is their only reasonable explanation.9
.
In 1999, Michael Lynch noted in "The Age and Relationships of the Major Animal Phyla" that:
Clarification of the phylogenetic relationships of the major animal phyla has been an elusive problem, with analyses based on different genes and even different analyses based on the same genes yielding a diversity of phylogenetic trees.20
.
Note: Both of the above two quotes by Woese and Lynch are also references in Jonathan Wells' controversial book, Icons of Evolution,
which originally alerted me to these references. I have reviewed these papers and added to the Woese quote
as compared to the portion originally quoted by Wells.
.
In 1999 Philippe and Forterre wrote an article entitled, "The rooting of the universal tree of life is not reliable" in which they made the following comments:
"The addition of new sequences to data sets has often turned apparently reasonable phylogenies into confused ones. . . In general, the two prokaryotic domains were not monophyletic with several aberrant groupings at different levels of the tree. Furthermore, the respective phylogenies contradicted each others, so that various ad hoc scenarios (paralogy or lateral gene transfer) must be proposed in order to obtain the traditional Archaebacteria-Eukaryota sisterhood."16
.
.
Another 1999 Science article by Stiller and Hall:
"A precipitous acceptance of such widespread LGT places evolutionary biologists in the untenable position of adopting an unfalsifiable hypothesis, at least in terms of the techniques of comparative sequence analyses that currently dominate the field of molecular evolution. Any phylogenetic pattern inferred from any given gene can be fit to some suitable mix of conventional intraspecies gene transmission and interorganismal genetic promiscuity. Thus, unless more reliable evidence is uncovered, the scientific method requires that we invoke the idea of ubiquitous LGT only as a last resort." 17
.
And another 1999 Science article by Doolittle:
"Each new prokaryotic genome that appears contains dozens, if not hundreds, of genes not found in the genomes of its nearest sequenced relatives but found elsewhere among Bacteria or Archaea." 18
In a 1999 paper by Ann Miller, from the Yale Department of Molecular Biophysics and Biochemistry, entitle, "The Evolution of Phylogenetic Classification: From 16S rRNA to the Genomic Tree."
"The 16S rRNA tree is not an organismal phylogenetic tree; it is a gene tree. To move towards organismal phylogeny, scientists began creating trees based on other proteins. In many cases, the other phylogenies do confirm the rRNA tree, but no one consistent phylogeny has emerged." 19
Then Kechris et. al., wrote:
"Phylogenies constructed on nitrogen fixation genes are not in agreement with the tree-of-life based on 16S rRNA but do not conclusively distinguish between gene loss and LGT hypotheses. Using a series of analyses on a set of complete genomes, our results distinguish two structurally distinct classes of MoFe nitrogenases whose distribution cuts across lines of vertical inheritance and makes us believe that a conclusive case for LGT has been made." 22
.
There is even suggestion that lateral gene transfer (LGT) may be fairly common between single-celled organisms and multicellular creatures. In a 2007 paper published in Science, Hotopp et. al., argue that there has been "widespread lateral gene transfer" between endosymbiotic bacteria and insects and nematodes. 21
Consistent hierarchies, at least for the earliest branches of the supposed "Tree of Life", are falling apart with additional evidence.When a given organism has hundreds of genes which none of its supposed nearest evolutionary relatives have, evolutionists are left in a very perplexing position.In order to maintain their theory they must propose, in an ad hoc non-falsifiable manner, that these differences were not the result of evolution from a common ancestor over time, but were in fact the result of lateral transfer of pre-evolved sequences.This messes the notion of nested hierarchies up very badly as far as its being a "science" is concerned.It is not science since it is not falsifiable.It is nothing more than "just so" story telling.
In this line, the work of Douglas Axe is also interesting and quite relevant. In his analysis of protein sequence space Axe suggests that changes to enzymes that are well away from the active can only tolerate very limited variability before experiencing a loss of selectable functionality of their original type. This seems to challenge the idea of continuous variability within sequence space, even for nominally closely related sequences. Because of this, Axe concludes:
"Contrary to the prevalent view, then, enzyme function places severe constraints on residue identities at positions showing evolutionary variability, and at exterior non-active-site positions, in particular. Homologues sharing less than about two-thirds sequence identity should probably be viewed as distinct designs with their own sets of optimizing features."23
But what about the higher branches that do show a more consistent nested hierarchical picture? Remember, "Phylogenetic incongruities can be seen everywhere in the universal tree, from its root to the major branchings within and among the various taxa to the makeup of the primary groupings themselves." And, this problem is becoming more and more significant as more and more is discovered about genetic sequences. Even as far back as 1998 it was known that there were serious problems within the highest branches of the "Tree of Life". Amazingly enough, a 1998 article entitled, "Molecules remodel the mammalian tree", Je Jong (in Trends in Ecology and Evolution) concluded:
.
"The wealth of competing morphological, as well as molecular proposals [of] the prevailing phylogenies of the mammalian orders would reduce [the mammalian tree] to an unresolved bush, the only consistent clade probably being the grouping of elephants and sea cows." 30
.
And, this major problem doesn't seem to have gotten any better over time. In 2009, Syvanen compared two thousand genes that are common to humans, frogs, sea squirts, sea urchins, fruit flies and nematodes. In theory, he should have been able to use the gene sequences to construct an evolutionary tree showing the relationships between the six animals. He failed. The problem was that different genes told contradictory evolutionary stories. This was especially true of sea-squirt genes. Conventionally, sea squirts—also known as tunicates—are lumped together with frogs, humans and other vertebrates in the phylum Chordata, but the genes were sending mixed signals. Some genes did indeed cluster within the chordates, but others indicated that tunicates should be placed with sea urchins, which aren't chordates. “Roughly 50 per cent of its genes have one evolutionary history and 50 per cent another." This led Syvanen to conclude: "We’ve just annihilated the tree of life."
Other scientists agree with the conclusions of the New Scientist article. Looking higher up the tree, a study published in Science tried to construct a phylogeny of animal relationships but concluded that, "Despite the amount of data and breadth of taxa analyzed, relationships among most [animal] phyla remained unresolved" (Link). Likewise, Carl Woese, a pioneer of evolutionary molecular systematics, observed that these problems extend well beyond the base of the tree of life:
.
"Phylogenetic incongruities [conflicts] can be seen everywhere in the universal tree, from its root to the major branchings within and among the various taxa to the makeup of the primary groupings themselves." (Link).
.
Likewise, in 2006, biologist Lynn Margulis wrote in her article, The Phylogenetic Tree Topples:
.
"Many biologists claim they know for sure that random mutation (purposeless chance) is the source of inherited variation that generates new species of life and that life evolved in a single-common-trunk, dichotomously branching-phylogenetic-tree pattern! Especially dogmatic are those molecular modelers of the ‘tree of life’ who, ignorant of alternative topologies (such as webs), don’t study ancestors." 25
.
Striking admissions of troubles in reconstructing the "Tree of Life" also came from a 2006 paper in the journal PLOS Biology entitled, Bushes in the Tree of Life. The authors acknowledge that, "A large fraction of single genes produce phylogenies of poor quality," observing that one study "omitted 35% of single genes from their data matrix, because those genes produced phylogenies at odds with conventional wisdom." The paper suggests that, "Certain critical parts of the [tree of life] may be difficult to resolve, regardless of the quantity of conventional data available." The paper even contends that, "The recurring discovery of persistently unresolved clades (bushes) should force a re-evaluation of several widely held assumptions of molecular systematics." 26
Then, Elie Dolgin, in a June, 2012 article in Nature reported that short strands of RNA called microRNAs are, "tearing apart traditional ideas about the animal family tree." Dartmouth biologist Kevin Peterson who studies miRNAs lamented, "I've looked at thousands of microRNA genes, and I can't find a single example that would support the traditional tree." According to the article, miRNAs yielded "a radically different diagram for mammals: one that aligns humans more closely with elephants than with rodents." Peterson put it bluntly: "The microRNAs are totally unambiguous ... they give a totally different tree from what everyone else wants." 31 A 2013 paper in Trends in Genetics reported that, "The more we learn about genomes the less tree-like we find their evolutionary history to be." 32
What is also interesting is that this information isn't entirely new - yet it is still treated by many with a great deal of surprise. Even as far back as 2000 Trish Gura argued, also in the journal Nature, that there appeared to be no consistent agreement between genetic phylogenies and those based on more traditional morphological characteristics:
.
"On one side stand traditionalists who have built evolutionary trees from decades of work on species' morphological characteristics. On the other lie molecular systematists, who are convinced that comparisons of DNA and other biological molecules are the best way to unravel the secrets of evolutionary history. … So can the disparities between molecular and morphological trees ever be resolved? Some proponents of the molecular approach claim there is no need. The solution, they say, is to throw out morphology, and accept their version of the truth. 'Our method provides the final conclusion about phylogeny,' claims Okada. Shared ancestry means a genetic relationship, the molecular camp argues, so it must be better to analyse DNA and the proteins it encodes, rather than morphological characters that can end up looking similar as a result of convergent evolution in unrelated groups, rather than through common descent. But morphologists respond that convergence can also happen at the molecular level, and note there is a long history of systematists making large claims based on one new form of evidence, only to be proved wrong at a later date." 27
So, evolutionary mechanisms are used to explain both hierarchical and non-hierarchical patterns.No matter how high up the tree this lack of hierarchy goes, the theory of evolution would still be used to explain the origin of such patterns. For focal problems in the tree between branches at higher levels, a change in mutation rate, or notions like convergence, divergence, or even lateral gene transfer are used. The fact is that the theory of evolution cannot be falsified by either a universal or a focal lack of nested hierarchy. Beyond this, the hierarchical classification method was first introduced by creationists, not evolutionists. So, to say that hierarchical patterns, when present, definitely support the the theory of evolution over intelligent design theory is erroneous. The theory of evolution does not predict hierarchical patterns more than does intelligent design theory.Again, nested hierarchies can be found all the time in human designs.
However, the death knell to this whole thing is the fact that most of these phylogenetic trees are based on functional genetic sequences.That messes everything up. Evolutionists would have a much stronger case if the sequences in question were actually neutral with regard to phenotypic function, but they aren't.That is why the notion of "pseudogenes" was so popular for such a long time - until recently when pseudogenes were actually found to be functional. What this means is that the differences are clustered or nested because of the different functional needs of different organisms in different environments.
Many types of functional proteins shared between very different creatures, like cytochrome c, are quite similar overall. In fact, certain key positions are highly conserved.The differences are also quite interesting in that they are maintained over thousands and even millions of generations.This means that most of the differences for such sequences are not neutral, but are indeed functional. In such a protein, that is otherwise so similar, it wouldn't take much to get to a new sequence if the new sequence was more functionally beneficial or "optimal".
Some argue that this doesn't happen because the different sequences are equally beneficial or "optimal" if applied to the same organism. That is basically arguing that the differences are not in fact functional different, but are actually neutral with respect to a functional optimum.Again, that makes no sense in light of the evidence that the differences, in addition to the similarities, are maintained over time.If this neutral argument were correct, then the distribution of sequences would be more randomly distributed. In other words, it would not be so neatly nested.
The evidence of functional maintenance over time is very strong evidence that those sequences that still appear to be somewhat "nested" are not so much the result of common ancestry as they are the result of various functional needs of different organisms in different environments.The more different the overall phenotype combined with the overall environment, the more different one can expect the individual sequences of a great many genes and proteins to be.And, this is pretty much what we find in real life.
Others have attempted to counter by suggesting that whales and dolphins should then be more similar to fish than to mammals since they live in the water instead of on land.Of course, the problem with this suggestion is that environment is not the only thing that plays into various genetic functional optimums. The other genes and the overall phenotype of the organism must also be considered.We should not expect the cytochrome c sequence of a dolphin to be the same a shark just because they both live in the same environment and eat many of the same things.Why? Because they have very different overall phenotypes.Also, we should not expect the cytochrome c sequence of a dolphin to be the same as that of a cow just because they are both classified as "mammals". Why?- Because they occupy very different environments and have a just a few differences in overall phenotype as well.
Even modern humans, when occupying different environments, will evolve different genetic sequences for various protein products that are actually functionally maintained over time due to various advantages that the differences provide in the different environments.There are many examples of this.And yet, when placed in the same environment, the differences quickly disappear in the offspring over time.Why?Because, there are indeed different optimal sequences when different overall phenotypes interact with different environments.
So again, I propose that the significant majority of differences between the cytochrome c of bacteria and humans are functional. They are not neutral.If the human sequence were put in a bacterium, it might survive ok, but it would not do as well.Over time, its offspring would rapidly evolve back the original more optimum sequence.
So far, the evidence is in fact far more consistent with the notion of common design with functional maintenance over time.It is starting to look a great deal like the books on my bookshelf or like the various types of cars on the highway - quite a few similarities combined with a great many distinct and isolated differences.
One data-point that might suggest common design rather than common descent is the gene “pax-6.” Pax-6 is one of those pesky instances where extreme genetic similarity popped up in a place totally unexpected and unpredicted by evolutionary biology. In short, scientists have discovered that organisms as diverse as jellyfish, arthropods, mollusks, and vertebrates all use pax-6 to control development of their very distinct types of eyes. Because their eye-types are so different, it previously hadn’t been thought that these organisms even shared a common ancestor with an eye. Evolutionary biologist Ernst Mayr explains the havoc wreaked within the standard evolutionary phylogeny when it was discovered that the same gene controlled eye-development in many organisms with very different types of eyes:
It had been shown that by morphological-phylogenetic research that photoreceptor organs (eyes) had developed at least 40 times independently during the evolution of animal diversity. A developmental geneticist, however, showed that all animals with eyes have the same regulator gene, Pax 6, which organizes the construction of the eye. It was therefore at first concluded that all eyes were derived from a single ancestral eye with the Pax 6 gene. But then the geneticist also found Pax 6 in species without eyes, and proposed that they must have descended from ancestors with eyes. However, this scenario turned out to be quite improbable and the wide distribution of Pax 6 required a different explanation. It is now believed that Pax 6, even before the origin of eyes, had an unknown function in eyeless organisms, and was subsequently recruited for its role as an eye organizer.
Typically, extreme genetic similarity is thought to mandate inheritance from a common ancestor, because the odds of different species independently arriving at the same genetic solution are exceedingly small. But if we require a Darwinian evolutionary scheme, such an improbable event is exactly what must have occurred. The observed distribution of genes like pax-6 demand extreme “convergent evolution” at the genetic level. Mayr tries to argue that such improbable examples of extreme genetic convergent evolution are not only acceptable, but common:
That a structure like the eye could originate numerous times independently in very different kinds of organisms is not unique in the living world. After photoreceptors had evolved in animals, bioluminescence originated at least 30 times independently among various kinds of organisms. In most cases, essentially similar biochemical mechanisms were used. Virtually scores of similar cases have been discovered in recent years, and they often make use of hidden potentials of the genotype inherited from early ancestors.
Mayr tries to explain away this extreme genetic convergent similarity by appealing to "hidden potentials of the genotype." Does this sound compatible with the kind of blind, unguided, and even random processes inherent in neo-Darwinian evolution? No. This sounds like a goal-directed process — intelligent design. (Link)
As Mayr suggests, there are other examples where genetic similarity appears in unexpected places. Biologically functional similarity that is thought to be the result of inheritance from a common ancestor is called "homology."
The concept of "homology" has been thrown into a crisis via observations, like those of Mayr, that the same genes control the growth of non-homologous body parts. Pax-6 is just one example. Another is the fact that the same gene controls the development of limbs in widely diverse types organisms that have wholly different types of limbs, where their common ancestor is not thought to have a common type of limb. The methodology used to infer homology was also challenged when it was discovered that different developmental pathways control the growth of body parts otherwise thought to be homologous. As the textbook Explore Evolution observes:
.
In sharks, for example, the gut develops from cells in the roof of the embryonic cavity. In lampreys, the gut develops from cells on the floor of the cavity. And in frogs, the gut develops from cells from both the roof and the floor of the embryonic cavity. This discovery—that homologous structures can be produced by different developmental pathways—contradicts what we would expect to find if all vertebrates share a common ancestor. … To summarize, biologists have made two discoveries that challenge the argument from anatomical homology. The first is that the development of homologous structures can be governed by different genes and can follow different developmental pathways. The second discovery, conversely, is that sometimes the same gene plays a role in producing different adult structures. Both of these discoveries seem to contradict neo-Darwinian expectations.
.
Perhaps this evidence is just the result of what Mayr called "hidden potentials of the genotype," or perhaps it contradicts neo-Darwinian expectations because neo-Darwinism is wrong. (Link)
A "few million years" might also be a problem for the resolution of mitochondrial D-loop sequences. Consider that the sequences used (two of them) to estimate the time of the most recent common ancestor (MRCA) between modern humans, Neandertals, and chimpanzees where each less than 400 base pairs in length (333bp and 340bp respectively).The mutation rate used by Krings et. al. was based on the a priori assumption that modern humans split off from chimps some "4-5 million years" ago.Based on this perhaps plausible, but indirect assumption, a substitution rate of 0.94 x 10-7 substitutions per site per year per lineage, was determined.Using this rate, the MRCA between humans and Neandertals was calculated to have lived about 465,000 years ago. The MRCA of modern humans was calculated to have lived around 163,000 years ago.And, the MRCA of chimps and bonobos was calculated to have lived around 2,844,000 years ago.3, 11, 13
Krings' figures are all fine and good except if we happen to come across a more direct measurement of mtDNA mutation rates. Consider the following work by Thomas Parsons published in the journal Nature Genetics:
"The rate and pattern of sequence substitutions in the mitochondrial DNA (mtDNA) control region (CR) is of central importance to studies of human evolution and to forensic identity testing. Here, we report a direct measurement of the intergenerational substitution rate in the human CR. We compared DNA sequences of two CR hypervariable segments from close maternal relatives, from 134 independent mtDNA lineages spanning 327 generational events. Ten substitutions were observed, resulting in an empirical rate of 1/33 generations, or 2.5/site/Myr. This is roughly twenty-fold higher than estimates derived from phylogenetic analyses. This disparity cannot be accounted for simply by substitutions at mutational hot spots, suggesting additional factors that produce the discrepancy between very near-term and long-term apparent rates of sequence divergence. The data also indicate that extremely rapid segregation of CR sequence variants between generations is common in humans, with a very small mtDNA bottleneck. These results have implications for forensic applications and studies of human evolution . . .
The observed substitution rate reported here is very high compared to rates inferred from evolutionary studies. A wide range of CR substitution rates have been derived from phylogenetic studies, spanning roughly 0.025-0.26/site/Myr, including confidence intervals. A study yielding one of the faster estimates gave the substitution rate of the CR hypervariable regions as 0.118 +- 0.031/site/Myr. Assuming a generation time of 20 years, this corresponds to ~1/600 generations and an age for the mtDNA MRCA of 133,000 y.a. Thus, our observation of the substitution rate, 2.5/site/Myr, is roughly 20-fold higher than would be predicted from phylogenetic analyses. Using our empirical rate to calibrate the mtDNA molecular clock would result in an age of the mtDNA MRCA of only ~6,500 y.a., clearly incompatible with the known age of modern humans. Even acknowledging that the MRCA of mtDNA may be younger than the MRCA of modern humans, it remains implausible to explain the known geographic distribution of mtDNA sequence variation by human migration that occurred only in the last ~6,500 years." 12
Several other more recent real time studies dealing with historical families have backed up Parson's findings. So, it seems as though more direct real-time measurements of mtDNA mutation rates show as much as a 20-fold higher mutation rate than that which was used by Krings et al. Now what does this mean - besides the obvious?
The sequences studied by Krings totaled 673 base pairs in length.According to the rate determined by Parsons, every single one of these base pairs would have changed more than twice in one million years and at least once in 400,000 years. Half of the base pairs would have mutated at least once in 200,000 years.And yet, humans are separated by only about 95 or so substitution differences from chimps?What is wrong with this picture? Each substitution difference (in a sequence some 673 base pairs in length) takes an average of 600 years to achieve. Taking into account that each lineage would build up substitution differences separately, in 600 years there would be around two substitution difference between two lineages. This seems to indicate that the common ancestor of humans and chimps lived some 30,000 years ago (not 4 to 8 million years ago as Krings et al., suggest - based on indirect methods). Modern humans, being separated from each other by an average of only 10 substitutions (according to Krings), appear to have a common ancestor living some 3,000 years ago.Modern Humans and Neandertals are separated by an average of only 35 substitutions.This seems to indicate a common ancestor living only some 10,000 years ago.
"It should be noted that molecular phylogenies are constructed on the basis of certain evolutionary assumptions. The tree that is presented is chosen from a forest of alternatives, typically on the assumption of maximum parsimony. That is, the tree that is selected is the one that reflects the least amount of presumed evolutionary change. But, if the assumption of maximum parsimony fails to fit the data, it can be jettisoned in favor of another."4In other words, any result can be accommodated by the theory by revising one or more of the underlying assumptions.
Even if a morphological phylogeny was matched closely by multiple molecular phylogenies, that would not prove that the groups in question descended from a common ancestor.The molecular differences could be linked to the morphological differences for some other reason. For example, all of the living organisms on this planet live in a relatively similar environment.All use the same water, breath the same air, and eat the same basic foods for building blocks and energy. Is it not reasonable to assume that a similar environment requires at least some similarities in the creatures that utilize it for survival?
Consider a world where plants utilized a different type of amino acid for protein metabolism than animals.This would mean that animals could not eat plants because the amino acids for one metabolic system would not necessarily work in the other system.The animals in such a world would be left with nothing to eat except for each other.The fact that creatures have many of the same or similar genes and proteins means that they are integrated with each other in their environment common environment (i.e., Earth).If they were not, they could not survive.Similar proteins and metabolic pathways are needed to utilize similar sources of food and energy.The various parts of organic life on this planet are interchangeable, not because of some random happenstance, but because of necessity - like Lego blocks.Building blocks made by some other company besides the Lego company would not likely "work" with Lego blocks (without prior knowledge of Lego blocks of course).
Nothing lives to itself.All living things are dependent upon other living things. If they were not molecularly and thus genetically compatible, nothing would survive very long.The "cycle of life" is dependent upon this fact. There would be no cycle if the basic building blocks of the creatures involved were not interchangeable with each other.Considering this need, it seems reasonable to assume that those creatures that share the most similar environments, body plans, and physiology would also have the most similar needs and thus the most similar genetic and molecular machineries.
Biologist Leonard Brand makes this point quite eloquently in the following excerpt:
"Anatomy is not independent of biochemistry. Creatures similar anatomically are likely to be similar physiologically. Those similar in physiology are, in general, likely to be similar in biochemistry, whether they evolved or were designed…An alternate, interventionist hypothesis is that the cytochrome c molecules in various groups of organisms are different (and always have been different) for functional reasons. Not enough mutations have occurred in these molecules to blur the distinct grouping evident.If we do not base our conclusions on the a priori assumption of megaevolution, all the data really tell us is that the organisms fall into nested groups without any indication of intermediates or overlapping of groups, and without indicating ancestor/descendant relationships."5
So, classification models of living things that are based on molecular similarities and differences are quite limited as far as their use as evidence of common ancestry beyond very recent times. Many differences that are maintained seem to be function based. Because of this, certain differences in sequences cannot be used as a "molecular clock" since natural selection fixes certain sequences based on functional needs so that random drift is not allowed. Beyond this, very different phylogenetic relationships can be hypothesized depending upon which sequence is subjectively chosen for analysis. These different trees are often outright incompatible with each other or, at best, inconclusive.
Maley, Laura E. and Charles R. Marshall. 1998. The Coming of Age of Molecular Systematics. Science Vol. 279 Issue 5350, p.505-506. ( Link to Full Text )
Patterson, Colin, and others. 1993. Congruence Between Molecular and Morphological Phylogenies. Annual Review of Ecology and Systematics 24:153-188.
Krings, M., Geisert, H., Schmitz, R., Krainitzki, H., and Pääbo, S. DNA sequence of mitochondrial hypervariable region II from the Neandertal type specimen. Evolution, Proc. Natl. Acad. Sci. USA, Vol. 96, pp. 5581-5585, May, 1999.
Hunter, Cornelius G. 2001. Darwin's God: Evolution and the Problem of Evil. Baker, Grand Rapids, MI.
Brand, Leonard. 1997. Faith, Reason, and Earth History. Andrews University Press, Berrien Springs, MI.
Davis, Percival and Dean Kenyon (editors). 1993. Of Pandas and People, second edition. Haughton Publishing Co., Dallas.
Denton, Michael. 1998. Nature's Destiny. Free Press, New York.
Ayala, Francisco J. 1978. The Mechanisms of Evolution. Scientific American 239:56-69.
Woese, Carl. 1998. The Universal Ancestor. PNAS June 9, 1998 vol. 95 no. 12 pp. 6854-6859: ( Link )
Elizabeth Pennisi, Is It Time to Uproot the Tree of Life? Science, vol. 284, no. 5418, 21 May 1999, p. 1305
Parsons, Thomas J. A high observed substitution rate in the human mitochondrial DNA control region, Nature Genetics vol. 15, April 1997, pp. 363-367
Krings M., Capelli C., Tschentscher F., Geisert H., Meyer S., von Haeseler A. et al. (2000): A view of Neandertal genetic diversity. Nature Genetics, 26:144-6.
JA King and RP Millar, Comparative aspects of luteinizing hormone-releasing hormone structure and function in vertebrate phylogeny, Endocrinology, Vol 106, 707-717 ( Link to Abstract )
Philippe H, Forterre P., "The rooting of the universal tree of life is not reliable." J Mol Evol. 1999 Oct;49(4):509-23. ( Link to Abstract )
John W. Stiller, Benjamin D. Hall, "Lateral Gene Transfer, Genome Surveys, and the Phylogeny of Prokaryotes" Science, Vol 286, Issue 5444, 1443 , 19 November 1999 ( Link to Text )
W. Ford Doolittle, Science 286, 1999
Ann L. Miller, "The Evolution of Phylogenetic Classification: From 16S rRNA to the Genomic Tree." Department of Molecular Biophysics and Biochemistry, New Haven, CT, 1999 ( Link to Text )
Michael Lynch, "The Age and Relationships of the Major Animal Phyla", Evolution, Vol. 53, No. 2, April 1999, pp. 319-325:
Julie C. Dunning Hotopp et. al., Widespread LateralGeneTransfer from Intracellular Bacteria to Multicellular Eukaryotes, Science 21 September 2007: Vol. 317. no. 5845, pp. 1753 - 1756
Katherina J. Kechris et. al., Quantitative exploration of the occurrence of lateral gene transfer by using nitrogen fixation genes as a case study, PNAS, June 20, 2006, vol. 103, no 25, p. 9584-9589. ( Link )
Douglas D. Axe, "Extreme Functional Sensitivity to Conservative Amino Acid Changes on Enzyme Exteriors," Journal of Molecular Biology, Vol. 301:585-595 (2000). | See also: Douglas D. Axe, "Estimating the Prevalence of Protein Sequences Adopting Functional Enzyme Folds," Journal of Molecular Biology, 1-21 (2004).
Graham Lawton, "Why Darwin was wrong about the tree of life," New Scientist (January 21, 2009). (Link)
Lynn Margulis, “The Phylogenetic Tree Topples,” American Scientist, Vol 94 (3) (May-June, 2006).
Antonis Rokas & Sean B. Carroll, "Bushes in the Tree of Life," PLOS Biology, Vol 4(11): 1899-1904 (Nov., 2006)
Masami Hasegawa, Jun Adachi, Michel C. Milinkovitch, "Novel Phylogeny of Whales Supported by Total Molecular Evidence," Journal of Molecular Evolution, Vol. 44, pgs. S117-S120 (Supplement 1, 1997)
Michael S. Y. Lee, “Molecular phylogenies become functional,” Trends in Ecology and Evolution, Vol. 14:177-178 (1999)
The mutation rate for humans is estimated to be about 2.2 x 10-9 mutations per base pair per year. With about 6.3 billion base pairs per genome, that works out to around 14 transmissible mutations per year per person.
By random chance alone in a *neutral* sequence we would expect to see 25% identity and 75% non-identity between two DNA sequences.We could achieve this "random look" with only 50% of each of two DNA sequences undergoing random mutation.So, how long would it take for 50% of a neutral 1000 character DNA sequence to get mutated?Well, it would take about 2.2 million years for each mutation or about 1.1 billion years for a random look.
But, what happens if the sequence is not functionally neutral?Well, now the population size, reproductive rate, and generation time comes into play.Given a population of say, 1 billion individuals, just about every character in our 1000-character sequence will get mutated every year in the population as a whole. A beneficial mutation will be spread throughout the population more or less rapidly depending upon the degree of its beneficial nature. Sex allows for the genetic recombination of the most advantageous mutations thus preferentially clustering them and making it appear that the random mutation rate was higher than it really was. Also, one mustn't forget about the oft touted "lateral gene transfer" theory, which is equivalent to sex in bacteria (and other creatures).
Nature Genetics 34, 264 - 266 (2003)
Published online: 22 June 2003; | doi:10.1038/ng1181
.
Convergent evolution of gene circuits
Gavin C Conant & Andreas Wagner
Convergent evolution occurs on all levels of biological organization, from organ systems to proteins. For instance, eyes and wings have evolved independently multiple times, and many aquatic vertebrates share a streamlined shape, despite their independent evolutionary origins1. On the smaller scale of proteins, lysozymes have been recruited independently for foregut fermentation in bovids, colubine monkeys and a bird2, 3. Antifreeze glycoproteins in antarctic notothenioids and northern cod (living at opposite ends of the globe) have independently evolved similar amino acid sequences4.
Convergent evolution is a potent indicator of optimal design. We show here that convergent evolution occurs in genetic networks. Specifically, we show that multiple types of transcriptional regulation circuitry in Escherichia coli and the yeast Saccharomyces cerevisiae have evolved independently and not by duplication of one or a few ancestral circuits.
J. J. Bull, M. R. Badgett, H. A. Wichman, J. P. Huelsenbeck, D. M. Hillis, A. Gulati, C. Ho and I. J. Molineux
Department of Zoology, Institute of Cellular and Molecular Biology, University of Texas, Austin, Texas 78712
Replicate lineages of the bacteriophage {phi}X 174 adapted to growth athigh temperature on either of two hosts exhibited high rates of identical, independent substitutions. Typically, a dozen or more substitutions accumulated in the 5.4-kilobase genome during propagation. Across the entire data set of nine lineages, 119 independent substitutions occurred at 68 nucleotide sites. Over half of these substitutions, accounting for one third of the sites, were identical with substitutions in other lineages. Some convergent substitutions were specific to the host used for phage propagation, but others occurred across both hosts. Continued adaptation of an evolved phage at high temperature, but on the other host, led to additional changes that included reversions of previous substitutions. Phylogenetic reconstruction using the complete genome sequence not only failed to recover the correct evolutionary history because of these convergent changes, but the true history was rejected as being a significantly inferior fit to the data. Replicate lineages subjected to similar environmental challenges showed similar rates of substitution and similar rates of fitness improvement across corresponding times of adaptation. Substitution rates and fitness improvements were higher during the initial period of adaptation than during a later period, except when the host was changed.
The following is an interesting exchange I had on Talk.Origins with a science teacher by the name of John Harshman (at least I think its interesting) regarding the observed pattern of living things. This pattern, generally speaking, forms what is known as a "nested hierarchy". John argues that this pattern, by itself, supports the theory of common descent (or modification of what came before over time) - even if it known that the key elements of the pattern required intelligent input. This is at least a fairly unique argument that I don't usually encounter in my debates. Perhaps some of you will find it interesting as well . . .
My comments are marked by double carats (> >) and no carats ( ) while John's are marked by a single carat ( > ):
> > Sean Pitman wrote:
> > Your efforts to presuppose limits on all intelligent designers, even
> > ones you do not know, reduces your hypothesis to a position of non-
> > falsifiability. Given the way you describe your position, it is true
> > by definition. It cannot be challenged, even in theory, because you
> > defined what a designer can and cannot do.
> John Harshman wrote:
> No, in fact I haven't. Consider this in a likelihoodist framework: A
> designer (hey, can I save typing by calling him "god" from now on?
> Thanks.) has a flat probability distribution of expected result,
> infinitely wide -- i.e. he could do anything. This means that the
> probability of any one outcome -- e.g. a nested hierarchy -- is
> arbitrarily close to zero. The likelihood of the data given the god
> model is almost nil. Then again, the distribution for common descent is
> sharply peaked; we strongly expect a nested hierarchy and little else.
> So the likelihood of the nested hierarchy data given the common descent
> model is quite high. In a likelihoodist framework we clearly pick common
> descent as an explanation of the data. Similar reasoning would produce
> similar results in bayesian or frequentist frameworks.
Sean Pitman wrote:
Let's say that we know the nested hierarchical pattern (NHP) was in fact designed, but we don't know the method of design. Given this scenario, you seem to be suggesting that, even given that ID produced the NHP, odds are the creative method chosen by the intelligent creator was common descent (CD)? - because only CD has a sharp likelihood peak given a NHP?
What is interesting here is that this notion is testable and it's outcome is not "almost nil" as you suggest. For example, give a bunch of people, from artists to housewives, a piece of paper and a pencil and tell them to sketch out various objects according to a NHP. Do you actually think that none of them will use any other method besides common descent to produce the NHP?
Not according to Michael Leyton, Dept. of Psychology, Rutgers University. In his book, Lyton argues that the "human perceptual system is organized as a nested hierarchy of symmetries." He goes on to argue that "architects exploit this psychological fact in the structure of their buildings" . . . and that the "same is true of painters, and of composers."
It seems only natural then that we humans tend to use NHP in our own creations without being told to do so and that we do not always use CD to produce our buildings, paintings, or other "compositions". In other words, the odds that a NHP, that is known to be designed, is the product of CD is not "essentially 100%" as you suggest.
> Or we could talk about specified information. A nested hierarchy is
> specified information. If we see a particular pattern that we expect to
> find resulting from X, we don't invoke some other process that has no
> expectation. The probability of getting that specified result from
> chance, or from an unpredictable god, are close to zero.
Not if you know that ID was *required* to produce the NHP. We know enough about the abilities of our own intelligence to know that we can easily skip the common descent steps and produce the NHP directly - de novo. In fact, this is often done in various creations that exhibit the NHP (as noted above).
> You actually use this reasoning yourself in other contexts. You only
> invoke god when convenient, and reject one when you think a natural
> model applies.
I wouldn't call this "convenient". I would call this a necessity. I invoke ID only when it seems to me that there is no other viable option. This is in fact the basis of the ID-only hypothesis - the same basis used by SETI scientists in their search for ID in the form of ET-produced "artifacts".
> Recently you claimed that the geological record is
> clearly the product of a catastrophic event. I said you couldn't rule
> out god creating the record. And your response was that since natural
> processes could explain the record, god was unnecessary.
What I reject is the notion that only ID could produce such a phenomenon. I do not reject the possibility that ID was involved. It is just that this notion cannot be adequately supported by the available data in this particular case.
> You reject god
> as an explanation purely because there is a natural explanation, because
> the probability of god producing a result that happens to mimic a
> natural process is, in your mind, very low.
That's not my reasoning at all. The probability of an intelligent agent mimicking a non-deliberate process of nature is not "essentially nil" as you suggest. We humans do it all the time. We make "natural gardens" and "natural rocks" to go in these gardens and paint natural scenes and produce the sounds of nature. We copy nature all the time - deliberately. So, unlike you, I do not reject the potential of ID for any phenomenon. What I reject is the notion that only ID could have done the job for certain phenomena - like the Grand Canyon or the geologic column. Other non-ID mechanisms could also do the job without my being able to tell the difference.
> And this reasoning doesn't
> come from a constraint put on god; quite the opposite: it comes from a
> total lack of constraint, which makes the probability of any particular
> result almost zero.
Nope.
> Similarly, you have already agreed that nested hierarchies are a
> predicted product of common descent;
Only given that non-deliberate natural processes could produce the various key differences in the various elements that make up the NHP.
> we have no need of god to explain
> that hierarchy just as we have no need of god to explain the
> stratigraphic record.
Not all NHP are created equal in that not all of them can be explained without the use of ID. For example, the NHP observed in certain architectural structures, paintings, and compositions require ID. They cannot be produced without ID. Such creations which demonstrate NHPs are demonstrably independent of the need for CD much of the time.
> Yet you reject common descent but accept stratigraphy. Why the
> difference? Simple, it's the elephant in the room that you won't
> mention. You have a prior template into which all conclusions must fit:
> biblical inerrancy. You know that common descent is false because
> Genesis says kinds were separately created. But you know strata weren't
> created because Genesis says (or is interpreted as implying) that the
> strata formed naturally from the Flood. All your argument is in service
> to that hidden agenda. And that's where the difference comes from.
> Nowhere else. Have the honesty to realize that.
I know that the key differences between the various "kinds" of creatures required ID. This is not true of stratigraphy. The various aspects of stratigraphy do not *require* ID. That's the difference.
This observation has nothing to do with the Bible. I'd be an IDist without the Bible. In fact, I thought that the ToE was quite reasonable for quite some time. It wasn't until after medical school when I was in the army that I discovered that the evolutionary mechanism simply didn't work beyond very low levels of functional complexity. It wasn't until then that I really started reconsidering the ToE.
< snip repetitive >
> > The difference between the nested pattern in living things and the
> > stratigraphic pattern is that all the key differences in the nested
> > pattern of life require ID.
>
> Irrelevant even if true, because we are talking about an aspect (the
> nested hierarchy itself) that we both agree doesn't require ID. Unless
> you are arguing for guilt by association, everything you say about this
> doesn't matter.
It is not irrelevant if true. All you have to do to see the relevance is ask a bunch of people to deliberately create something that expresses a NHP and see if they use CD as a mechanism. You see, when ID is known to be involved, it is also known that NHPs are often produced without the need for CD.
< snip >
> > This is quite different from the "tree of life" pattern where just
> > about all the branches of the tree require intelligent input. Given
> > that intelligent input is in fact required to produce most of the
> > differences in the tree, the overall pattern is in fact the result of
> > ID.
>
> Sorry, that doesn't follow. It's like saying that because apples grow on
> trees, and an apple pie is mostly apples, then apple pies must grow on
> trees.
Nope. It is like saying that when a NHP is a known product of ID, common descent is often bypassed to get to the final creation faster. You see, intelligent minds can progress through all the CD steps to the end product within the mind - without having to produce each step separately. The end product that exhibits a NHP can be produced right away without the need to *physically* use the process of CD.
< snip >
> > One is known to require intelligent design to produce the pattern
> > while the other does not require intelligent design to produce the
> > pattern.
>
> Again, you make this work only by confusing what pattern we're talking
> about. No intelligent design is required to produce the pattern of
> nested hierarchy, as you yourself admit.
Again, not all phenomena that exhibit a NHP can be produced without the input of ID. Those creations the exhibit NHP, and are also *known* to require ID, can be and often are produced without the use of CD.
To see the rest of this discussion see the following link at Talk.Origins (Link).
A second debate concerning the problem of convergent evolution is as follows:
On Apr 11, 2:52 pm, John Harshman <jharshman.diespam...@pacbell.net> wrote:
>
> > I never said that convergence was "miraculous".What I said was that
> > it can be expected when the genetic sequence is under the influence of
> > natural selection.This viral mutation experiment wasn't just a
> > general trend to more GC vs. AT in a sequence.Numerous identical
> > mutations were achieved in specific nucleotide sites.Of course, this
> > convergence made it impossible to "recover the correct evolutionary
> > history."
>
> Such convergence under selection is possible if there is only one set of
> changes that can be selected. But that's seldom if ever likely. I don't
> have access to the complete Genetics paper. Do you?
Yes, I do. Here are a few of the key comments of Bull et. al.:
MOLECULAR sequences are routinely used to infer evolutionary histories across the entire spectrum of life. Underlying these uses of molecular data is the assumption that convergent evolution is statistically unimportant - that identical, independent changes in different lineages are not common enough to obscure the true historical signal (DOOLITTL1994; HEDGES and MAXSON 1996). However, the impact of convergent molecular evolution on phylogenetic reconstruction is difficult to evaluate because the true phylogeny is almost never known and thus cannot be used to identify errors in the phylogeny estimated from molecular data. This potential effect on phylogenetic reconstruction is complicated by the fact that not enough is known about the evolutionary causes of convergence to predict where it might be a problem
A rarity of convergence in molecular evolutioin is not obviously controversial, as it can be reconciled with both the selectionist and neutralist paradigms. Under neutrality, most substitutions are stochastic, soconvergence is merely accidental. If instead, most substitutions are considered adaptive, convergence is likewise unexpected unless organisms face the same selective environment, and then only if they have few alternative pathways of adaptation to that environment. Experimental systems provide a direct way of assessing the magnitude of convergent evolution. Most obviously, they avoid the circularity of using inferred phylogenies to infer properties of molecular evolution that themselves influenced the reconstruction. Beginning and end points of the evolutionary process can be assessed directly to avoid uncertainty about which changes occurred. . .
Fitness improved substantially in all evolved lines (Figure 2). The initial fitness of ancestor A was low on both hosts at 42 degrees, whereas, by the 11th day, fitnesses of isolates within each primary lineage had improved at least 4000-fold ( -12 doublings/ hr). . . It appears that fitness improvement is greater early than late except when the phage is placed in a new environment.
Rates of nucleotide substitution showed a pattern similar to rates of fitness change (Table 1 ) . All isolates differed from A by at least 12 and upt o 26 substitutions, and a 27-base deletion was observed in SC1. [The same deletion was observed previously and did not appear to have an obvious effect on viral replication (MULLER and WELLS 1980), but caused a 50% increase in eclipse rate ( INCARDONA and MCJLLER1 985) .]On a per day basis, the number of substitutions was relatively uniform across the five primary lineages and even on the extended lineages SC1 and SC2, ranging from 12 to 15 per 11 days. Extended lineages S1+ and C2+ showed a significantly lower rate of substitution from that of the primary lineages (six and seven changes per 22 days, P < 0.01 ).
Despite the low per-genome rate of substitution ( 0.2-0.5%), many of the same substitutions occurred independentlyin two or more lineages (Tables 1 and 2). In addition, some of the changes that accumulated inthe primary lineages were reversed in extended lineages,further contributing to the convergence (Table2 deals specifically with convergences). These levels of convergence are difficult to reconcile with random models of substitution, which assume that convergence is coincidental because of high rates of substitution. . . The fact that, with one exception, all sites experiencing substitutions in more than one lineage were also substituted for the same base further contributes to the deviation between this process and randomness.
Although some convergence was observed between most pairs of lineages, the highest levels of convergence were typically observed among lineages grown on the same host. . .
The facts that S3 was grown from its own isolated plaque of A and that SCl and SC2 were grown from separate isolated plaques of S1, combined with the high levels of convergence between these lineages and the (other) primary lineages, indicates that convergence was not an artifact of peculiarities inherent to the A lysate. . .
Erroneous phylogenetic reconstruction: The convergent substitutions interfered with an accurate reconstruction of the true history of these lineages.Considering first the six-taxon tree that contains A and the five primary lineages (Sl, S2, S3, C1, C2), the maximum likelihood reconstruction united all five evolved lineages on a branch from ancestor A, as if all five shared a common ancestor more recently than A (Figure 3). The tree further divided the three S. typhimurium evolved lineages (Sl, S2, S3) from the two E. cob evolved lineages (Cl, C2). Although some structure is to be expected when using small samples to reconstruct star phylogenies, the strong clustering of phage isolates evolved on the same host suggests more than just sampling error. This intuition is supported statistically: the likelihood for the true tree was significantly worse than for the estimated tree, indicating that the true tree is rejected (at P ~0.01, Figure 3) . This erroneous reconstruction is not simply due to our incorporation of ancestor A into the analysis (the ancestor would typically be absent in nonexperimental studies) : reconstruction without ancestor A also yields an estimate that is significantly different from a star phylogeny ( P < 0.01; analysis
not shown).
The estimated changes were mapped on the reconstruction in Figure 3 to consider how much of the parallelism would be detected when the true phylogeny is not known. When a reconstruction is compromised by parallelism, the parallelism should be underestimated because it is often interpreted as common ancestry. A total of 26 parallel substitutions were observed across 16 sites in the five primary lineages (a single parallelism occurs with the second independent origin of a change, a second parallelism with the third independent origin, and so on), Only nine of the 26 (35% ) parallelisms were detected in the reconstruction, at nine of the 16 sites. The underestimation is thus appreciable. Reconstruction of the full set of 10 taxa is likewise compromised (Figure 4). As with the six-taxon tree, the true phylogeny is rejected at the 1% level. . .
Convergence may indicate the beneficial effects of some substitutions. . . A substitution process driven by selection is also consistent with the observed excess of missense substitutions. The ratio of missense to silent changes is significantly higher than expected fort his spectrum of mutations (P < 0.01, expectations calculated empirically by randomly assigning the observed nucleotide changes across all sites in the genome, comparing the number of substitutions silent in all reading frames to substitutions causing missense changes in at least one reading frame) . The inference of selection from an excess of missense changes is indirect, however, and may not provide a true measure of the fraction of sites that were selected. Furthermore, silent substitutions may not be neutral: position 500 evolved silent substitutions in all three S. typhimurium lineages and reverted to the ancestral state in one of the SC lineages.
Discussion:
Our study may be the first to show that an erroneous phylogenetic reconstruction results from convergent molecular evolution (DOOLIITLE 1994; HEDGES and MAXSON 1996; but see BEINTEMA et al. 1977) . Two manifestations of the problems caused by convergence were as follows: (i) the number of changes since common ancestry of the lineages was underestimated, and (ii) lineages grown on the same host were grouped together in the reconstruction. The errors in reconstruction were not an artifact of sampling just a small portion of the genome, because the entire genome sequence was used. Furthermore, the estimated histones were statistically significant deviations from the true histories.
How general is a high rate of convergence? Virtually all phylogenetic trees based on molecular data reveal homoplasy, so some level of convergence is common if not universal (PEACOCK and BOULTER 1975; BEINTEMA et al. 1977;R OMERO-HERRERA et al. 1979; SANDERSON and DONOGHU1E9 96; WELLS1 996). Should we then extrapolate from the present study and conclude that convergence commonly causes mistakes in reconstruction? Not yet. First, a ubiquity of convergence is not, by itself, a problem for reconstruction. . .
Three properties of the design may have led to the high rate of convergence, all of which are atypical of what is thought to apply to most organisms:
1. Replicate lineages were exposed to nearly identical
2. Strong, mass selection was operating on very large
3. The replicate lineages started with virtually identical
We can offer plausible arguments for why all three properties might contribute to convergence: (i) similar environments create similar selective pressures, which in turn favor similar adaptations (a hypothesis consistent with the observed phylogenetic clustering of lineages grown on the same host) ; (ii ) the array of beneficial mutations with large effects, which will be those favored under strong selection, is much more limited than those with small effects; and (iii) the fitness effect of a mutation is genome dependent, so when the same genome is subjected to the same selection, the same mutations will be favored. [This is basically my same argument in a nutshell]. . .
Finally, CUNNINGHAM et al. (1997) reported multiple, independent origins of deletions and stop codons in experimental lineages of T7 subjected to mass selection with infrequent bottlenecks. The selective factor in that study was grossly similar to that in the YIN ( 1993) study (loss of a T7 function was beneficial to phage growth ), except that selection was applied to large populations without spatial structure. Thus, in comparing these three T7 studies, the nature of selection (property 2) may explain the different levels of convergence.
Despite our hesitance in regarding these øX 174 results as typical of other systems, one should not be too quick to dismiss convergence as a problem in phylogenetic reconstruction. Indeed, the phenomenon known as “long branch attraction†results when high levels of substitution create enough convergence to impair reconstruction (FELSENSTEIN, 1978). The fact that two thirds of parallelisms in the øX 174 primary lineages would have been misdiagnosed had we not known their true history cautions that the extent of convergence in other systems may be underestimated whenever the true history is unknown. . . .
In an interesting application of convergence, MOLLA et al. ( 1996) used parallelisms among human immunodeficiency virus (HIV) isolates to identify substitutions that were likely to be (and later were shown to be) important in drug resistance. The present case even extends those precedents in that the adaptation has occurred to broad environmental agents (temperature, host, and perhaps media) rather than to a specific challenge such as a drug, and convergence is genome-wide rather than confined to a single gene.
> >>> This conclusion is supported by David Hillis in his paper published in
> >>> the journal "Phylogeny" in 1999, "SINEs of the Perfect Character".
> >> The journal is PNAS; no journal named "Phylogeny" exists to my knowledge.
>
> >>> Hillis writes:
> >>>"What of the claim that the SINE/LINE insertion events are
> >>> perfect markers of evolution (i.e., they exhibit no homoplasy)?
> >>> Similar claims have been made for other kinds of data in the past, and
> >>> in every case examples have been found to refute the claim. For
> >>> instance, DNA-DNA hybridization data were once purported to be immune
> >>> from convergence, but many sources of convergence have been discovered
> >>> for this technique. Structural rearrangements of genomes were thought
> >>> to be such complex events that convergence was highly unlikely, but
> >>> now several examples of convergence in genome rearrangements have been
> >>> discovered. Even simple insertions and deletions within coding regions
> >>> have been considered to be unlikely to be homoplastic, but numerous
> >>> examples of convergence and parallelism of these events are now known.
> >>> Although individual nucleotides and amino acids are widely
> >>> acknowledged to exhibit homoplasy, some authors have suggested that
> >>> widespread simultaneous convergence in many nucleotides is virtually
> >>> impossible. Nonetheless, examples of such convergence have been
> >>> demonstrated in experimental evolution studies."
> >>> Of course, Hillis goes on to point out that certain types of mutations
> >>> are usually non-homologous and non-convergent.However, the fact
> >>> remains that functional elements and even some types of non-functional
> >>> genetic elements do experience convergence.
> >> This again is not the sort of convergence you have in mind. A SINE
> >> insertion is another single mutation. There may indeed by hot spots of
> >> SINE insertion.
>
> > This wasn't thought to be true until recently.
>
> There were hints of it as much as 8 years ago. But so?
It was a creationist prediction before 8 years ago - -I predicted it myself.
> >> Now what you're talking about, which Hillis refers to
> >> here as "widespread simultaneous convergence in many nucleotides", may
> >> have examples. But you have not apparently presented any. Hillis
> >> references the Bull et al. paper, on which he was a co-author.
>
> > That is a valid example.
>
> If so, you have exactly one example so far. And I'm not sure it fits
> your needs anyway.
Several examples are listed in the Bull paper.
> >> By the way, do you know what Hillis says in the very next sentence after
> >> your quote? Here: "All of these sources of data remain useful and
> >> important for the inference of phylogeny. Therefore, the presence of
> >> homoplasy is not, in itself, terribly problematic, as long as
> >> appropriate statistical assessments are made of the inferences based on
> >> the data."
>
> > Yes, as long as the "appropriate statistical assessments are made" and
> > the interference of NS is not too great.That's a big if.
>
> In fact it's not a big if. You don't know what you're talking about and
> appear unwilling to listen to those (like Hillis) who do. It's
> interesting that Hillis is your authority when it suits your purposes,
> but you feel free to ignore his claims when that suits. This is yet more
> of the selective use of science by creationists that I began this thread
> to demonstrate.
As one of the co-authors will Bull, Hillis also concludes:
"Despite our hesitance in regarding these øX 174 results as typical of other systems, one should not be too quick to dismiss convergence as a problem in phylogenetic reconstruction. Indeed, the phenomenon known as “long branch attraction†results when high levels of substitution create enough convergence to impair reconstruction (FELSENSTEIN, 1978). The fact that two thirds of parallelisms in the øX 174 primary lineages would have been misdiagnosed had we not known their true history cautions that the extent of convergence in other systems may be underestimated whenever the true history is unknown. . ."
It is my suggestion that you are in fact being "too quick to dismiss convergence as a problem in phylogenetic reconstruction."The same thing is true of Hillis and Bull.There is a strong desire to build these phylogenetic trees and to put a great deal of weight on them, but this might be rather problematic given such experimental results.
> >> So what exactly are you trying to get at? It appears that you are
> >> claiming phylogenetic inference to be impossible, or at least so
> >> unreliable as to be meaningless. Yet nobody in this discussion other
> >> than you and Schwabe seems to think so, and with good reason.
>
> > When it comes to the assumption of evolutionary relationships that
> > developed over the course of many millions of years, the problem of
> > convergent and parallel mutations can become quite an issue.Yes, you
> > and Hillis and others are quite optimistic that this problem can be
> > reliably overcome.I don't think so.All bets are off when NS is in
> > play over long periods of time.
>
> Such is your unsupported claim. But it should be fairly easy to see that
> this can't be true. First, many sequences and sites are evolving
> neutrally.
That is certainly your assumption.However, many sequences that were once thought to be nothing more than evolutionary "junk" are now being shown to be functional and under the pressures of NS.So, don't be so hasty to call a particular sequence functionally "neutral".
> Second, even if some sequences are under selection, it's
> absurd to suppose that this leads to widespread convergence in detail,
> and particularly that it results in a hierarchical pattern of
> convergence.
This is exactly what Hall et. al. demonstrated - - convergence in detail at the DNA level.
> Third, it's even more absurd to suppose that all parts of
> the genome should be subject to identical patterns of convergence.
Oh really?
"Another study showed that the genetic distance among flowering plants is much greater than that among mammals, even though flowering plants [are thought to] have evolved for similar amount of time as mammals (Huang, 2008a). The genetic distance between two subpopulations of medaka fish that had diverged for ~ 4 million years is 3-fold greater than that between two different primate species (humans and chimpanzees) that had diverged for 5-7 million years (Kasahara et al., 2007). The genetic distance measured on genealogical timescales (< 1 million years) is often an order of magnitude greater than that on geological timescales (> 1 million years) (Ho and Larson, 2006), suggesting that genetic distance measured in evolutionary time is independent of actual mutation rate measured in real time. . .
It is a self-evident intuition that simpler systems/machines can tolerate more variations/choices in building blocks. The more complex the system, the more restriction would be placed on the choice of building blocks. A one-story house can be build by all varieties of bricks but only the stronger ones among them can qualify for a 100-story building because the weaker ones cannot withstand the weight of a 100-story building. The number of choices of different materials for constructing a toy bicycle is much greater than that for a space shuttle. . . Thus, simpler organisms with low epigenetic complexity can tolerate more variations in DNAs or have higher genetic diversity. Genetic diversity is defined here as genetic distance or dissimilarity in DNA or protein sequences between different individuals or species. . .
It is well known that sequence regions conserved in simple organisms are often also conserved in complex organisms. Sequence regions not conserved in complex organisms are also often not conserved in simple organisms. This explains the fact as illustrated in Table 1 that a comparison of fish (with a hypothetical maximum diversity of 60%) and human (with a hypothetical maximum diversity of 10%) should result in a dissimilarity of 60% equaling the maximum diversity of fish, rather than 70%.
This notion that the maximum genetic diversity of a simple kind of organism determines and is about the same as the maximum distance between the simple organism and the later appearing complex organisms can be illustrated by the example of cytochrome c. The maximum diversity in this protein sequence is about 70% difference within bacteria, for example, between Bordetella parapertussis and Paracoccus Versutus. The maximum distance between bacteria and mammals is about 65% difference, such as between Bordetella parapertussis and Pan troglodytes (chimpanzees). Within fungi, the maximum diversity is about 40% difference, for example, between Aspergillus oryzae and Yarrowia lipolytica. The maximum distance between fungi and mammals is about 43% difference, such as between Aspergillus oryzae and Pan troglodytes. Within arthropods, the maximum diversity is about 24% difference, for example, between Drosophila melanogaster and Tigriopus californicus. The maximum distance between arthropods and mammals is about 25% difference, such as between Drosophila melanogaster and Pan troglodytes. . .
In a survey of 40 randomly picked genes, I found 36 (90%) that show the expected macroevolution pattern where human is more related to bird, less to frog, and still less to fish. In contrast, only 19 (48%) show more identity between the two fishes than between human and bird. Depending on which gene is used as clock, the time of divergence between the two fishes would vary from 91 to 420 million years (Table 2). In fact, I employed the molecular clock method to derive an average time of divergence using these 40 genes by calibrating against the fossil divergence time between human and bird (310 MyBP). However, I obtained an obviously incorrect time (417+/-172 MyBP) that is more than two fold greater than the actual time as indicated by the fossil record. As a positive control to show that my method is similar to those of others, I derived a mean time of divergence between human and amphibians and found it to be similar to that obtained by others (Kumar and Hedges, 1998).
Apparently, some of the subspecies split or microevolution is not equivalent to the changes in macroevolution, but the Neo-Darwinian hypothesis treats them the same. . .
It is well known that mutation rate from pedigree analysis on genealogical timescales is often an order of magnitude or more greater than mutation rate from phylogenetic analysis over geological time (Ho and Larson, 2006). Thus, phylogenetic diversity or distance over geological time is uncoupled from actual mutation rate observed on genealogical timescales. It suggests that actual mutation rates are often fast enough for most organisms to reach a maximum cap in genetic distance over geological timescale.
> I will note again what you seem to be unwilling to face, that you are
> being inconsistent. One day you agree that there is an objectively
> determinable nested hierarchy of life, and the next you claim that any
> such hierarchy is an illusion. What exactly are you really trying to
> say? How does it support your underlying claims, whatever they may be?
I've already explained my position to you before - several times.I do in fact recognize a general nested hierarchical pattern to the tree of life.I just think that this NHP may not mean what you think it means.The assumed common ancestry could be more a function of common functionality and resulting limitations on divergence as well as parallel mutations and convergence than common descent.
You are the one who says that it is essentially statistically impossible for an intelligent designer to produce a particular natural pattern, such as a NHP, because there are so many other options available.Well John, as I've pointed out to you before, this argument is completely vacuous given that human designers incorporate specific natural patterns in various designs all the time.But, you argue, the NHP seen in the tree of life is not used in human designs to any significant degree.Even if you were correct with that argument, and you aren't, it's a different argument than your initial statistical argument.Your original argument that the odds of using a particular natural pattern are essentially zero is demonstrably false because it has no real application outside of your assumed limitation from using a "natural" NHP.It is therefore a meaningless argument.
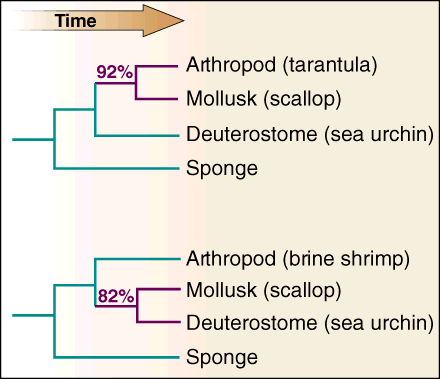 The article goes on to present similarities and differences in 18s rRNA sequences which show that mollusks (scallops) are more closely related to deuterostomes (sea urchins) than arthropods (brine shrimp). Of course, this is not too surprising. Intuitively, a scallop seems more like a sea urchin than a shrimp. So, the 82% correlation between the scallop and sea urchin is not surprising. However, in this light it is surprising is that a tarantula (also an arthropod) has a 92% correlation with the scallop. Here we have two different arthropods, a shrimp and an tarantula. How can a scallop be much more related to one type of arthropod and much less related to the other type of arthropod? This troubling thought led the authors of the Science article to remark:
The article goes on to present similarities and differences in 18s rRNA sequences which show that mollusks (scallops) are more closely related to deuterostomes (sea urchins) than arthropods (brine shrimp). Of course, this is not too surprising. Intuitively, a scallop seems more like a sea urchin than a shrimp. So, the 82% correlation between the scallop and sea urchin is not surprising. However, in this light it is surprising is that a tarantula (also an arthropod) has a 92% correlation with the scallop. Here we have two different arthropods, a shrimp and an tarantula. How can a scallop be much more related to one type of arthropod and much less related to the other type of arthropod? This troubling thought led the authors of the Science article to remark: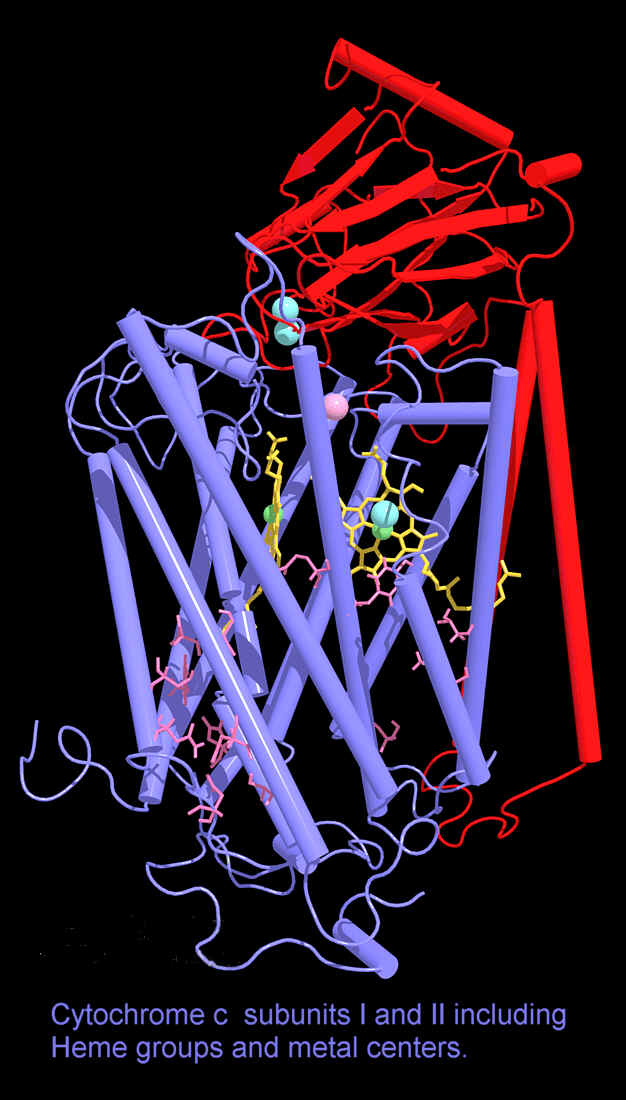 Calcitonin (lowers blood calcium levels in animals) is another protein commonly used to determine phylogenies.
Calcitonin (lowers blood calcium levels in animals) is another protein commonly used to determine phylogenies. The question is, then, if bacteria can achieve such relatively rapid neutral genetic drift, why are they not more wide ranging in their cytochrome c sequences? It seems that if these cytochrome c sequence differences were really neutral differences, that various bacterial groups, colonies, and species, would cover a rather large range of possible cytochrome c sequences - potentially to include that of mammals. Why are they then so uniformly separated from all other "higher" species unless the different cytochrome sequences are different for functional reasons? that they are statically different due to the various different functional needs of creatures that inhabit different environments?
The question is, then, if bacteria can achieve such relatively rapid neutral genetic drift, why are they not more wide ranging in their cytochrome c sequences? It seems that if these cytochrome c sequence differences were really neutral differences, that various bacterial groups, colonies, and species, would cover a rather large range of possible cytochrome c sequences - potentially to include that of mammals. Why are they then so uniformly separated from all other "higher" species unless the different cytochrome sequences are different for functional reasons? that they are statically different due to the various different functional needs of creatures that inhabit different environments?